
AI enthusiast, ML researcher and amatuer skier ⛷️
I'm Max, a machine learning reseacher based in Munich 🇩🇪. I currently pursue a PhD at the University of Glasgow 🏴 where I research and develop technologies that aim to ensure privacy in machine learning applications.
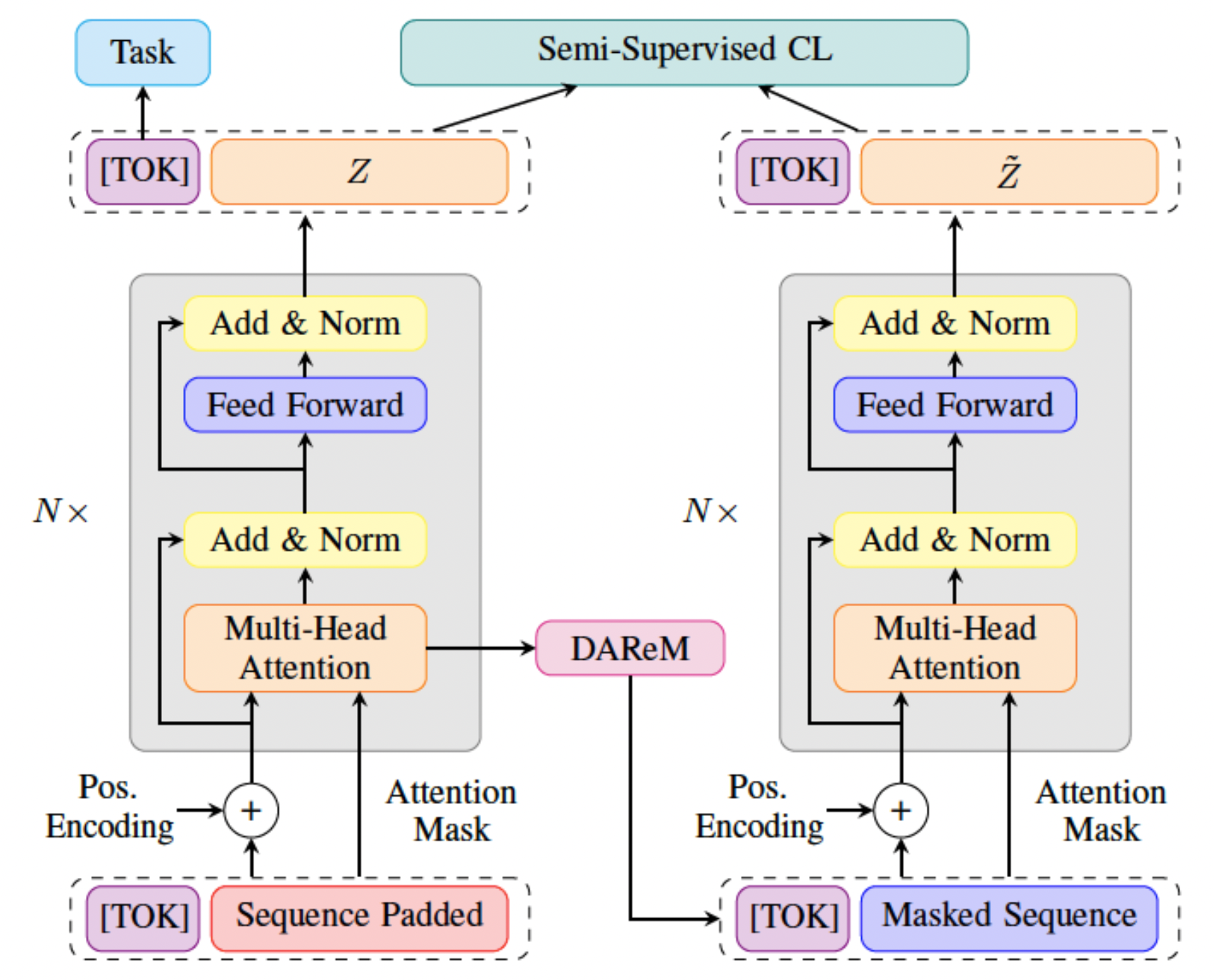
STaRFormer: Semi-Supervised Task-Informed Representation Learning via Dynamic Attention-Based Regional Masking for Sequential Data
NeurIPS 2025
Forstenhäusler, M., Külzer, D., Anagnostopoulos, C., Parambath, S., Weber, N.
Understanding user intent is essential for situational and context-aware decision-making. Motivated by a real-world scenario, this work addresses intent predictions of smart device users in the vicinity of vehicles by modeling sequential spatiotemporal data. However, in real-world scenarios, environmental factors and sensor limitations can result in non-stationary and irregularly sampled data, posing significant challenges. To address these issues, we propose STaRFormer, a Transformer-based approach that can serve as a universal framework for sequential modeling. STaRFormer utilizes a new dynamic attention-based regional masking scheme combined with a novel semi-supervised contrastive learning paradigm to enhance task-specific latent representations. Comprehensive experiments on 56 datasets varying in types (including non-stationary and irregularly sampled), domains, sequence lengths, training samples, and applications demonstrate the efficacy of STaRFormer. We achieve notable improvements over state-of-the-art approaches.
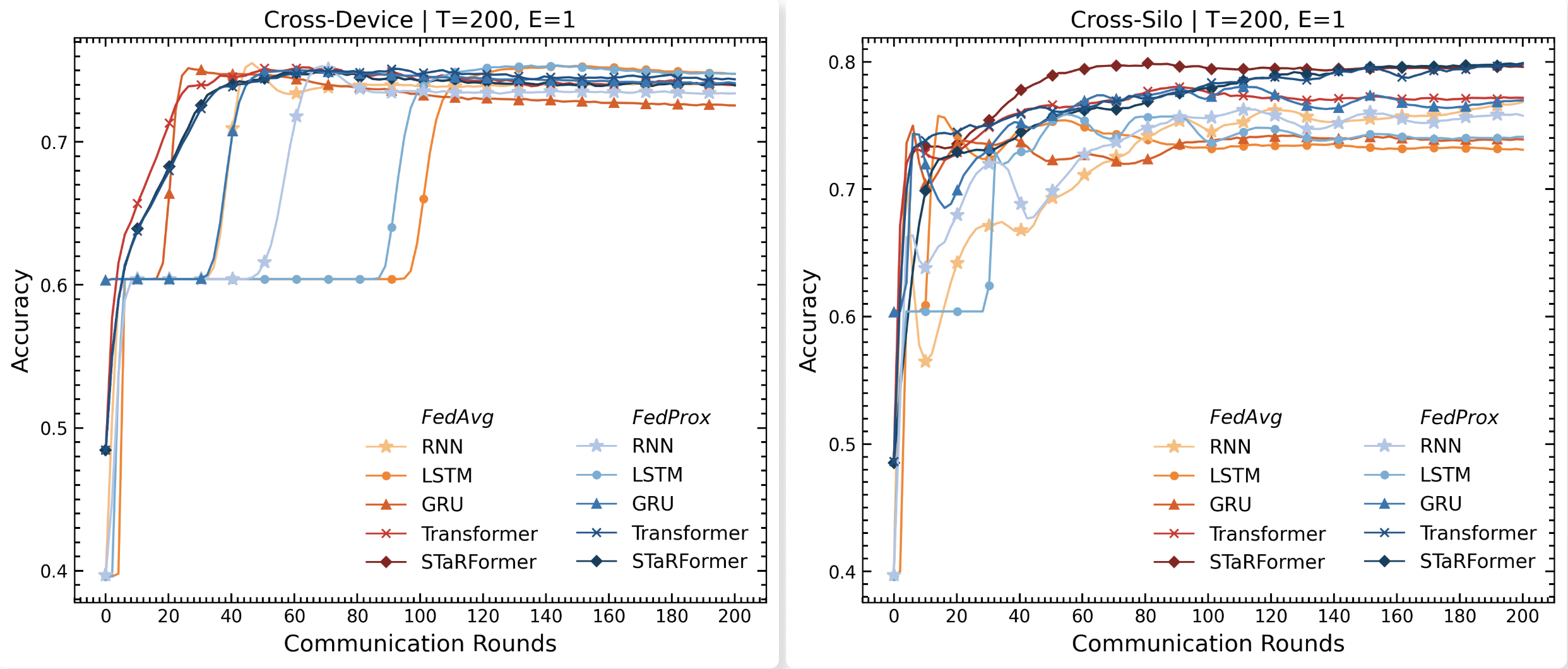
Leveraging Federated Learning for Decentralized Semi-Supervised Task-Informed Representation Learning on Sequential Data
IEEE ICDCS 2025
Forstenhäusler, M., Anagnostopoulos, C., Parambath, S., Külzer, D., Weber, N.
Over the past decade, advancements in machine learning, particularly large language models like OpenAI’s Chat-GPT, have heightened public awareness about artificial intelligence. The growing awareness has led to increased expectations for intelligent products that enhance user experience, often requiring the collection of substantial amounts of potentially sensitive data. However, data protection legislation such as the EU AI Act or GDPR restricts the collection and storage of sensitive user data in real-world applications, making it difficult for organizations to aggregate large datasets across users or institutions, in addition to substantial transmission costs associated with storing the data in the first place. Federated Learning offers a promising paradigm to address these issues by enabling decentralized learning via data minimization, thereby also re- ducing communication overhead. To evaluate its effectiveness, we explore a real-world sequential classification task to learn the intent of smart device users within confined areas of a vehicle. Utilizing an anonymized real-world dataset sourced from BMW’s car fleet and employing a novel sequential modeling approach, STaRFormer, we analyze two federated learning architectures, cross-device and cross-silo, and assess two federated aggregation algorithms for various baseline models. The results demonstrate that STaRFormer outperforms baseline models in the cross-silo case and delivers competitive results in the cross-device case, highlighting its efficacy in decentralized sequential data modeling.
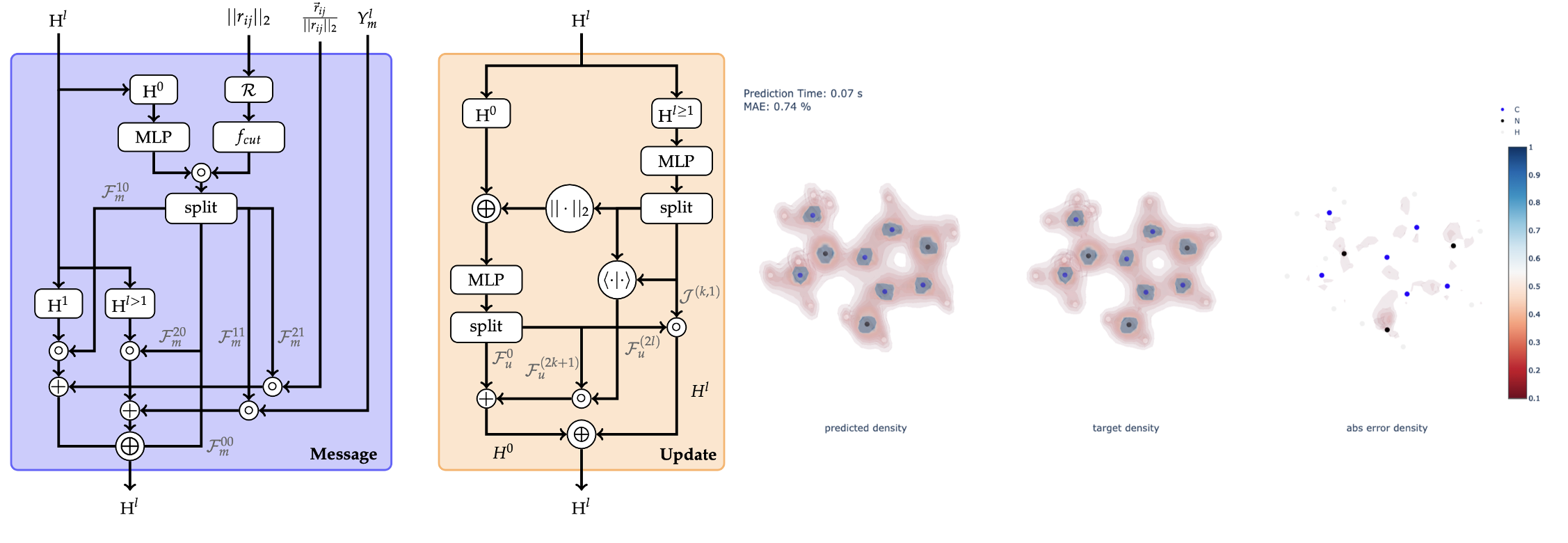
Physics-Informed Geometric Deep Learning for Molecular Property Prediction
TUM Library
Forstenhäusler, M.
In recent years, data-driven methods have become increasingly appealing to predict and accelerate chemical property prediction and molecular dynamics. Leveraging graph representations, graph neural networks have become the method of choice for this task. Initially, these models aimed to incorporate invariance to permutations and translations to align with the laws of physics. Nevertheless, subsequent work demonstrated that it is crucial to not only account for invariances but also to address rotational equivariance of molecular structures. Most commonly, performance benchmarking of machine learning models in this space occurs on the QM9 and MD17 datasets. However, most recently, it was shown that electron densities inherently contain more information than energy and other common properties of these benchmarks. Hence, this work emphasizes predicting a molecule’s electron density from its 3-dimensional atomistic representation via a data-driven approach. To predict densities, this study provides an extension to the QM9 dataset by computing density target values, i.e., densities coefficients, via density functional theory. Moreover, this work extends two algorithmic methodologies, a message passing and an attention-based methodology, to maintain rotational equivariance while including an arbitrary number of type-l features per irreducible representation, which is a prerequisite to predict and compute the electron density. Initial benchmarking revealed that the message passing model performs considerably better than the attention-based model. In fact, the message passing model, benchmarked on common public datasets, performs on par with state-of- the-art methodologies. Likewise, the electron density predictions achieve excellent results with mean absolute errors of 0.30%. Even when training on a small subset of data, decent errors of 1.37% on the test dataset are achievable. In summary, two different methodologies of equivariant machine learning models are introduced and benchmarked on common datasets. In addition, the scope of available properties within the QM9 dataset is extended to allow the prediction of information-rich electron densities.
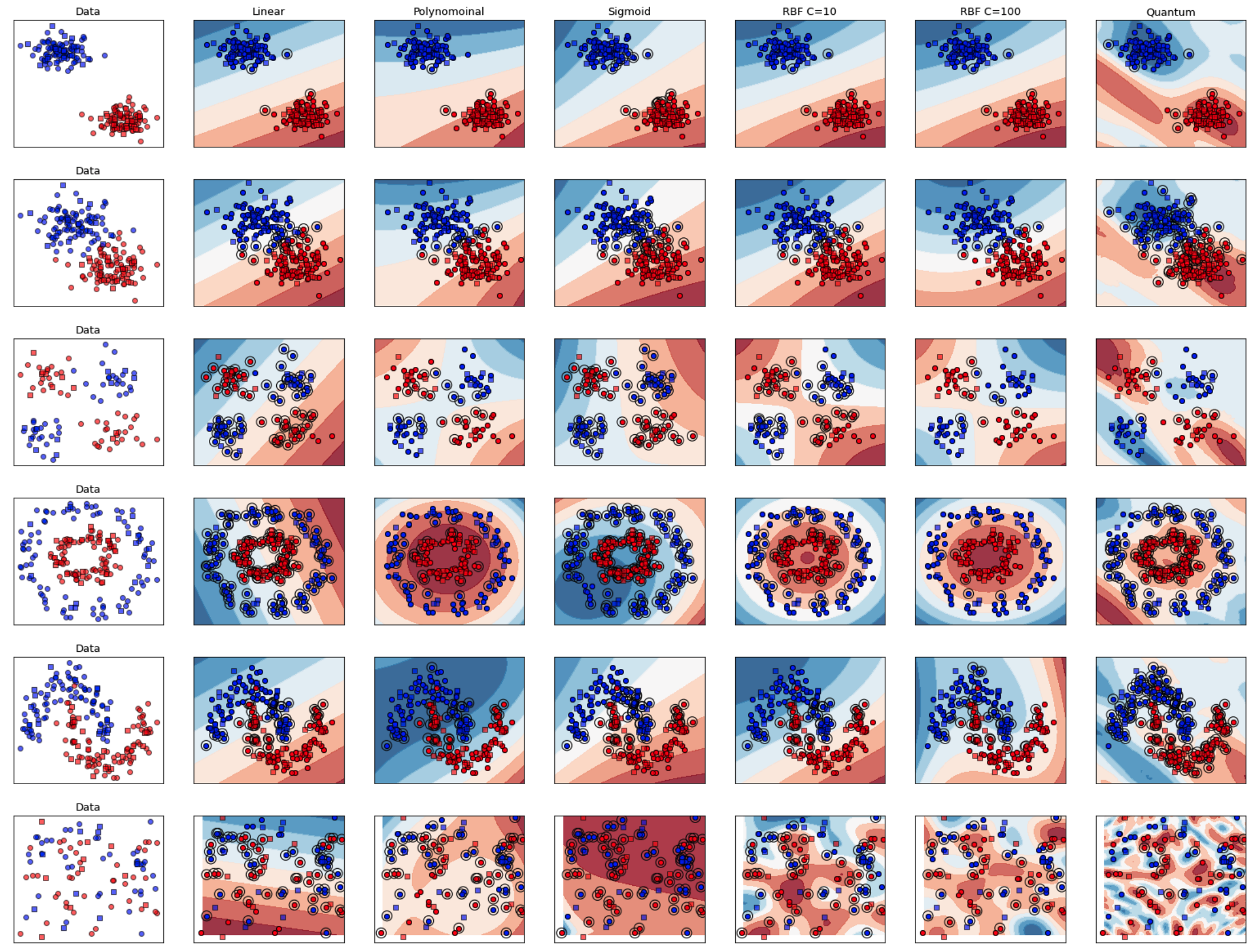
Quantum Support Vector Machines
Forstenhäusler, M.
As part of a Seminar at during my M.Sc. at TUM, I experimented with the implementation of Quantum Support Vector Machines and Classical Suport Vector Machines.
Work Experience

BMW
PhD candidate in Federated Learning
03/2024 - Present

Caltech
Visiting Researcher
11/2022 - 10/2023
BMW
Working Student in Machine Learning
03/2024 - 10/2022
TUM.ai
Software Dev Team Lead
06/2021 - 05/2022

George Washington University
Undergraduate Research Assistent
05/2018 - 05/2020

Robert Bosch GmbH
Intern - EPS
05/2019 - 08/2019